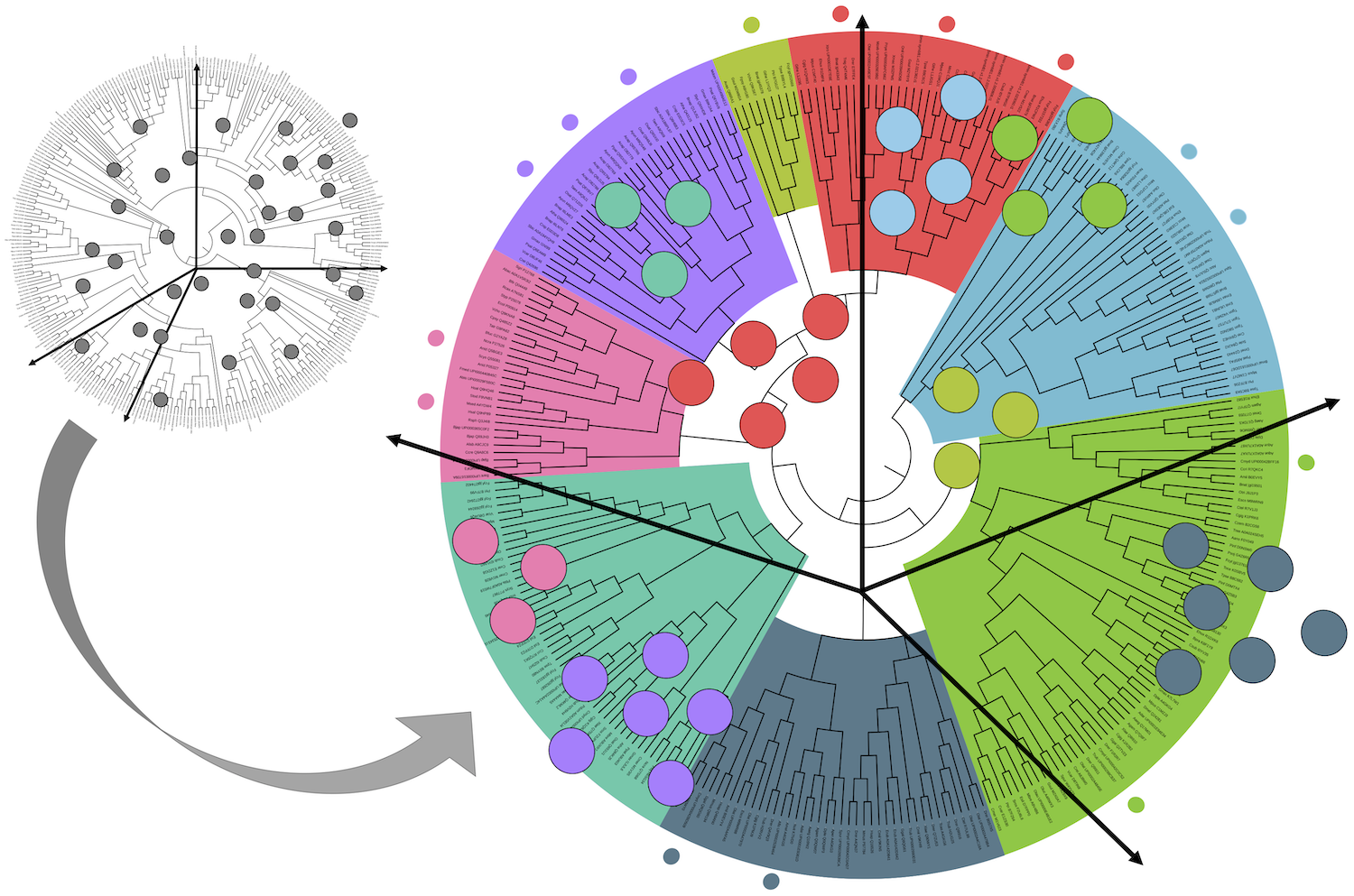
Predicting species-specific mutation effects is a major challenge in genetics. Using diverged sequences, we learn protein sequence landscapes, which predict tolerated mutations in a species like E. coli and capture interactions between mutations.
See our article in Nature Communications.
|
|
|
|
|
Towards predicting new mutations in SARS-CoV-2: in the very early stages of an emerging viral outbreak, can we predict new mutations in the proteins of the virus which might lead to its future variants? The "Statistical Genomics and Biological Physics" team proposes a computational approach based on a single viral genome infecting humans and pre-existing viral genomes infecting other species.
For more information see:
|
Gene duplication is a major source of functional diversification. However, the co-existence of two paralogues with similar biochemical properties but diverging functions can lead to potentially detrimental competition between the duplicates. This phenomenon has been named “paralogue interference”. In a recent article published in "Frontiers in cellular infection and microbiology, the "genetics networks" team of LCQB has addressed this question in the human pathogen Candida glabrata. They showed that evolution selected mutations which decreased competition between two, potentially interfering, transcription factors, thus allowing the emergence of the particular modes of regulation for respiration and iron homeostasis observed in extent Saccharomycetaceae species.
Link to the article
|
We provided crucial insights to the molecular mechanism through which HBV infects cells. A fusion peptide in preS1 and the human protein-disulfide isomerase ERp57 are involved in HBV membrane fusion process.
Link to the article
|
To avoid deleterious misfolding of proteins, the assembly of multiprotein complexes is tightly controlled and can either occur co-translationally in the cytoplasm or as a spatially restricted event by targetting ribosomes at particular subcellular locations. In an article recently published in Molecular Cell, Benoit Palancade's team (Institut Jacques Monod) showed that both phenomenon are at play in the biogenesis of the nucleopore, one of the largest multiprotein complex in the cells. The "genetics networks" team from LCQB contributed to this work by conducting genome-wide analyses of the interactions between nucleoporins (key nucleopore subunits) during translation
Link to the article
|
|
|
|
|
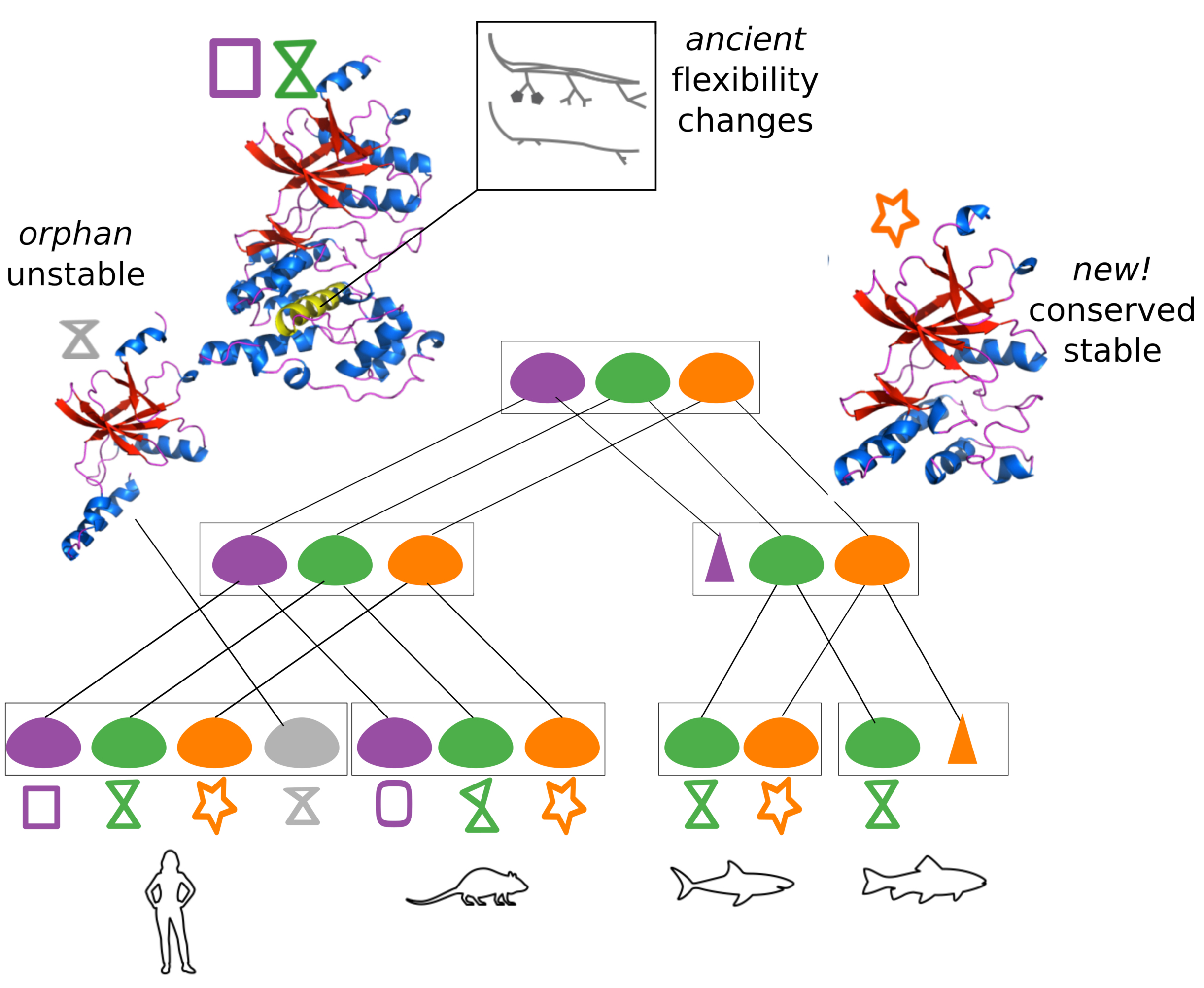
We propose PhyloSofS, the first automated tool to reconstruct plausible evolutionary scenarios explaining a set of observed transcripts, and to generate 3D molecular models of the protein isoforms. We apply it to the JNK family (60 transcripts, 7 trees) to identify AS events of ancient origin and relate their functional outcome with changes in the protein dynamics. We also show that PhyloSofS can help identify new potential therapeutic targets.
Link to the paper
|